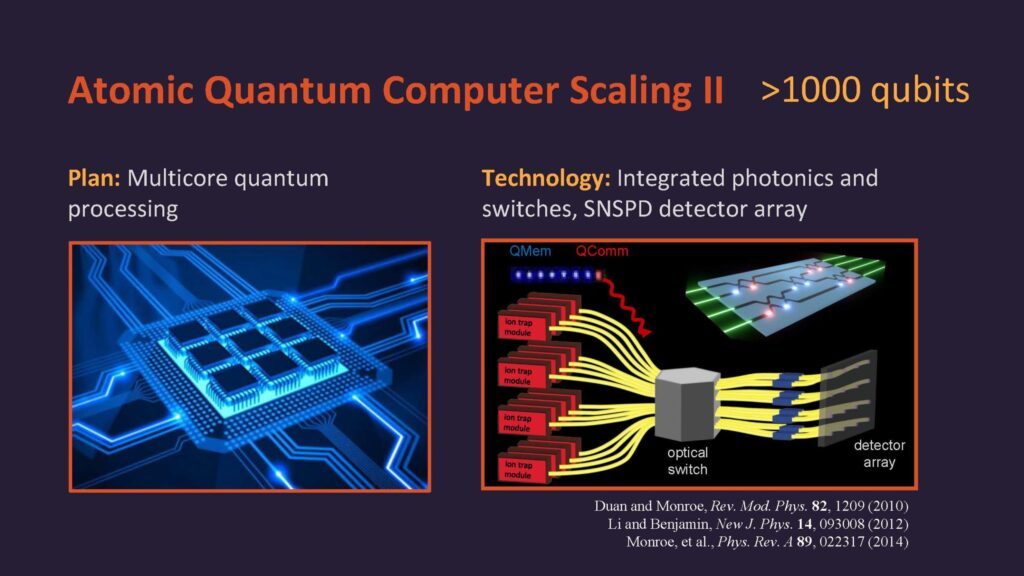
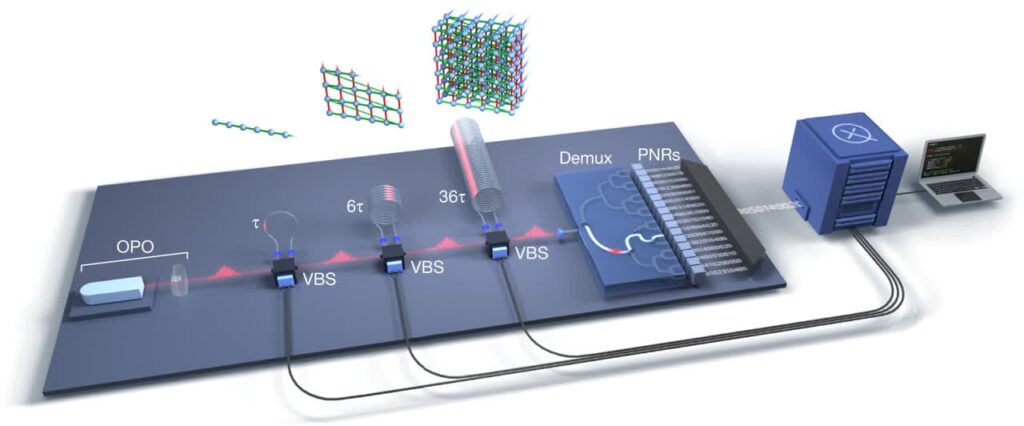
رایانه کوانتومی از مهمترین فناوریهای در حال توسعه محسوب میشود که سرمایهگذاریهای هنگفتی در مراکز تحقیقاتی معتبر دنیا بر روی آن صورت میگیرد. به طور کلی، رایانه کوانتومی ماشینی است که از ویژگیهای مکانیک کوانتومی مانند برهمنهی کوانتومی و درهمتنیدگی برای انجام محاسبات و پردازش اطلاعات استفاده میکند. استفاده از این ویژگیها منجر میشود تا رایانه کوانتومی محاسبات را با سرعت بیشتر و فضای محاسباتی کمتری نسبت به رایانه کلاسیک انجام دهد. محاسبات در رایانه کوانتومی با اجرای الگوریتمهای کوانتومی انجام میگیرد. الگورریتم کوانتومی مشابه الگوریتم کلاسیک یک مسئله را گام به گام حل میکند، با این تفاوت که از ویژگیهای کوانتومی بهره میبرد. الگوریتم کوانتومی در سادهترین شکل آن مجموعهای از دروازههای کوانتومی متوالی است که بر روی یک حالت اولیه معین اثر میکند و پس از اندازهگیریهای مربوطه بر روی حالت نهایی، جواب یک مسئله معین را با احتمال بسیار بالا به دست میدهد. الگوریتم کوانتومی با سرعت بسیار زیاد و در مدت زمان کم قادر به حل مسائلی است که الگوریتم کلاسیک ازحل آنها عاجز است. با این وجود، برای اجرای الگوریتم کوانتومی به صدها یا شاید هزاران کیوبیت نیاز است. از سوی دیگر، خطاهایی که در حین انجام الگوریتم رخ میدهند نیز باید تصحیح شوند.
امروزه الگوریتمهای کوانتومی در حوزههای مختلفی مانند جستجو میان دادهها، رمزنگاری، بهینهسازی، حل معادلات خطی، طراحی مواد، تحقیقات دارویی، مدلسازی واکنشهای شیمیایی و فیزیکی و شبیهسازی مولکولهای شیمیایی مورد استفاده قرار میگیرند. از این رو، با توجه به کاربرد فراوان الگوریتمهای کوانتومی، بررسی و مطالعه آنها از اهمیت بسزایی برخوردار است.
تعیین ساختار الکترونی مولکولها قلب شیمی محاسباتی
یکی از مهمترین کاربردهای رایانه کوانتومی در حوزه شیمی و به طور خاص تعیین ساختار الکترونی مولکولها است که از اهمیت بسیار فراوانی در حوزه شیمی محاسباتی و شیمی کوانتومی برخوردار است، به گونهای که از آن به عنوان قلب شیمی محاسباتی تعبیر میشود. از مهمترین اهداف تعیین ساختار الکترونی مولکولها، محاسبه انرژی حالت پایه مولکولی (Molecular Ground State Energy) است که نقش مهمی در بدست آوردن اطلاعات در مورد ساختار پایدار مولکول، ویژگیهای طیفسنجی مولکول، مکانیسم و سرعت واکنشهای شیمیایی ایفا میکند. با این وجود، به دلیل وجود همبستگیهای الکترونی در هامیلتونی مولکولی، تعیین ساختار الکترونی مولکولها به ویژه با افزایش اندازهی سیستم و تعداد الکترونها با چالش جدی محاسباتی همراه است. در نتیجه برای حل مسئله انرژی حالت پایه از روشهای تقریبی مانند Hartree-Fock (HF) و یا روشهایی با دقت بالاتر مانند Full Configuration Interaction (FCI) استفاده میشود.
در سالهای اخیر مطالعات تئوری و تجربی برای حل مسئله انرژی حالت پایه مولکولی با استفاده از الگوریتمهای کوانتومی انجام گرفته است. از الگوریتمهای کوانتومی پیشرو که در تعیین ساختار الکترونی مولکولها به کار میرود میتوان به الگوریتم VQE (Variational Quantum Eigensolver) اشاره کرد که از جمله الگوریتمهاي ترکیبی کوانتوم-کلاسیک محسوب میشود. نخستین مطالعه تجربی انجام شده در این حوزه، تخمین انرژی حالت پایه یون هلیوم هیدرید HeH+ بر روی رایانه کوانتومی با استفاده از الگوریتم VQE است که در سال 2014 در مجله Nature Communications به چاپ رسیده است. از آن زمان تاکنون این الگوریتم نقش بسزایی در استفاده از رایانه کوانتومی در تعیین ساختار الکترونی مولکولها ایفا میکند. به طور کلی در الگوریتم VQE، ویژهمقدار کمینه هامیلتونی مولکولی (انرژی حالت پایه مولکولی) بر اساس اصل تغییر (Variational Principle) در مکانیک کوانتومی تخمین زده میشود. در این الگوریتم، حالت اولیه سیستم با توجه به مسئله مورد نظر با روشهای مختلف مانند Unitary Coupled Cluster فراهم میشود که یک حالت پارامتري است. سپس، این حالت پارامتري که ansatz نامیده میشود به یک رایانه کوانتومی فرستاده میشود و اندازهگیري کوانتومی بر روی آن انجام میگیرد. در ادامه، نتایج اندازهگیري که تابعی از پارامترهاست به یک رایانه کلاسیک ارسال میشود. رایانه کلاسیک با انجام بهینهسازي، یک دسته پارامتر جدید بازتولید میکند که این پارامترها به رایانه کوانتومی بازگردانده شده و این حلقه تا یافتن پارامتر بهینه و کمینه شدن انرژی مولکول تکرار میشود. نکته حائز اهمیت این است که از آنجایی که در این الگوریتم از رایانه کلاسیک نیز استفاده میشود، تعداد کیوبیت مورد نیاز برای شبیهسازی کوانتومی کاهش مییابد.

رایانه کوانتومی، رویای شیمی محاسباتی
به طور رایج، از روشهای شیمی محاسباتی که مبتنی بر شیمی کوانتومی است، برای محاسبهی انرژی حالت پایه و ساختار الکترونی مولکولها استفاده میشود که با افزایش تعداد ذرات، فرایند شبیهسازی زمانبر خواهد بود و نیز ممکن است انحرافاتی از نتایج تجربی مورد نظر مشاهده شود. به همین دلیل استفاده از الگوریتمهای کوانتومی برای فرایند شبیهسازی جهت افزایش سرعت و دقت فرایند شبیهسازی پیشنهاد شده است. با این وجود، با افزایش تعداد الکترونها در ساختار مولکولها و نیاز به تعداد کیوبیت بالاتر در فرایند شبیهسازی کوانتومی (بر اساس تعداد الکترونها و اوربیتالها در دیاگرام اوربیتال مولکولی)، این حوزه از فناوری محاسبات کوانتومی به دلیل محدودیت در تعداد کیوبیت در دسترس با چالش روبرو است. از همین رو، مطالعات تئوری و بعضاً تجربی انجام شده در این حوزه محدود به مولکولها و ساختارهای شیمیایی نسبتا ساده مانند مولکولهای هیدوژن و آب است. اگر چه روشهایی برای کاهش تعداد کیوبیت مورد نیاز برای شبیهسازی کوانتومی مولکولهای شیمیایی ارائه شده است، با این وجود به نظر میرسد در آینده نه چندان دور با افزایش تعداد کیوبیت در دسترس، امکان تعیین ساختار الکترونی ساختارهای شیمیایی پیچیدهتر بر روی رایانه کوانتومی فراهم خواهد شد.